G100
version gromacs/2021.2
The first benchmark is the Pure MPI of the large system of 2136412 atoms (Ribosome in water), by means of the classic MD simulation with the time step of 4fs. The Performance is shown in Graphic 1 in yellow, while in grey is the ideal curve performance.
Table 1. Performance
| n nodes | n cores | ns/day |
| 0,25 | 12 | 1,534 |
| 0,5 | 24 | 2,695 |
| 1 | 48 | 4,863 |
| 2 | 96 | 10,547 |
| 4 | 192 | 20,100 |
| 8 | 384 | 35,623 |
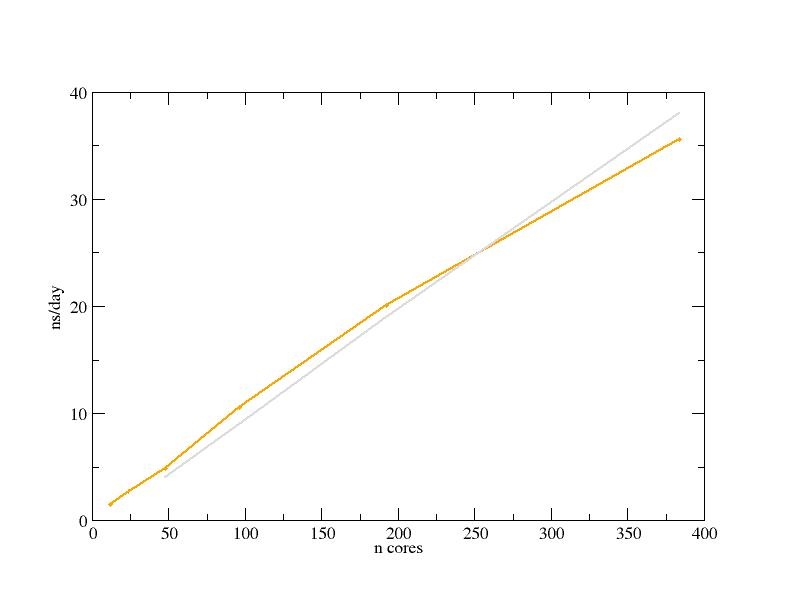
Graphic 1: the Gromacs performance (simulation time in ns/day) is reported vs. the increasing number of cores
The effect of mixed MPI+Threads parallelization scheme is shown in Graphic 2, where the number of MPI processes * Threads is kept constant to the maximum available (64) for each node. These data suggest to use a number of Threads between 4 and 16 (and therefore a number of MPI processes between 16 and 4) in order to achieve the best performance.
Graphic 2: the Gromacs performance (simulation time in ns/day) is reported vs. the increasing number of Threads at a constant number of MPI processes * Threads = 64

RUN SINGLE MD GMX 4.6
mdrun=$(which mdrun_bgq)
exe="$mdrun -s remd_0.tpr -ntomp 4"
runjob -n 4 --ranks-per-node 4 --env-all : $exe
© Copyright 2