hostname: login.marconi.cineca.it
early availability: 27/06/2016
start of production (A1 - Broadwell): 04/07/2016 Starting from September 26th, 2018 the activity on Marconi-A1 has been stopped.
start of production (A2 - Knights Landing): 04/01/2017 Starting from January, 2020 the activity on Marconi-A2 will be stopped.
start of production (A3 - Skylake): 07/08/2017
start of production (A3+ - Skylake): 23/01/2018
start of production (A3++ - Skylake): xx/10/2018
Marconi is the new Tier-0 system, co-designed by Cineca and based on the Lenovo NeXtScale platform, that substitutes the former IBM BG/Q system (FERMI). MARCONI, based on the next-generation of the Intel® Xeon Phi™ product family alongside with Intel® Xeon® processor E5-2600 v4 product family, offers the scientific community a technologically advanced and energy-efficient high performance computing system.
This achievement represents the first step of the Italian infrastructure development plan put forward by the Cineca governing bodies, aimed at supporting scientific research. The global plan entails an investment of Euro 50 million in two phases. The first, just started, will make available a computational power of about 20Pflop/s and a data storage capacity of more than 20 petabytes, which will go into production, reaching completion in the second half of 2017. The second phase will start during 2019, with a final goal to increase available computing power to approximately 50 to 60 Pflop/s by 2020.
The system, logically named ‘MARCONI’, has been designed to be gradually completed in about 18 months, between mid 2016 and beginning 2018, according to a plan based on a series of updates:
- A1: a preliminary system going into production in July 2016, based on Intel® Xeon® processor E5-2600 v4 product family (Broadwell) with a computational power of 2Pflop/s.
- A2: a new section has been added at the start of 2017, equipped with the next-generation of the Intel Xeon Phi product family (Knights Landing), based on a many-core architecture, enabling an overall configuration of about 250 thousand cores with expected additional computational power of approximately 11Pflop/s.
- A3: finally, in July 2017 - January 2018, this system reaches a total computational power of about 20Pflop/s making use of the future generation Intel Xeon processors (Sky Lakes).
This supercomputer takes advantage of the new Intel® Omni-Path Architecture, which provides the high performance interconnectivity required to efficiently scale out the system’s thousands of servers.
A high-performance Lenovo GSS storage subsystem, that integrates the IBM Spectrum Scale™ (GPFS) file system, is connected to the Intel Omni-Path Fabric and provides data storage capacity.
The progressive development of the Marconi system allows use of state-of-the-art processor technology, enabling an extremely high-performance system but still with a ‘green’ soul. One of the parameters of the project developed by the Cineca team is in fact to gradually increase the computational power up to 50Pflop/s without exceeding, at any stage, the limit of 3MWatt power consumption (see here the full https://wiki.u-gov.it/confluence/display/SCAIUS/Marconi+press+release+-+English).
System Architecture
Architecture: Intel OmniPath Cluster
Internal Network: Intel OmniPath Architecture 2:1
Disk Space: 17PB (raw) of local storage
Login nodes: 8 Login (3 available for regular users). Each one contains 2 x Intel Xeon Processor E5-2697 v4 with a clock of 2.30GHz and 128 GB of memory. Login nodes are shared between three partitions: A1 (BDW), A2 (KNL) and A3 (SKL). The three partitions are served by a single SLURM server.
In January 2020 the A2(KNL) partition has been upgraded to a new nVIDIA accelerated architecture. The MARCONI2 cluster is described in a separate document (UG3.1.1)
System A1 (Broadwell) - out of production since September 26th, 2018 Model: Lenovo NeXtScale Racks: 10 |
|
|---|

System A2 (Knights Landing) - out of production since January, 2020 Model: Lenovo Adam Pass Racks: 50 Peak Performance: 11 PFlop/s |
|---|

System A3 (Skylake) Model: Lenovo Stark Racks: xx |  |
|---|
Marconi Network
Network type: Intel Omnipath, 100 Gb/s. At the time of setup, MARCONI was the largest Omnipath cluster in the world.
Network topology: Fat-tree 2:1 oversubscription tapering at the level of the core switches only.
Core Switches: 5 x OPA Core Switch "Sawtooth Forest", 768 ports each.
Edge Switch: 216 OPA Edge Switch "Eldorado Forest", 48 ports each.
Maximum system configuration: 5(opa) x 768 (ports) x 2 (tapering) → 7680 servers.
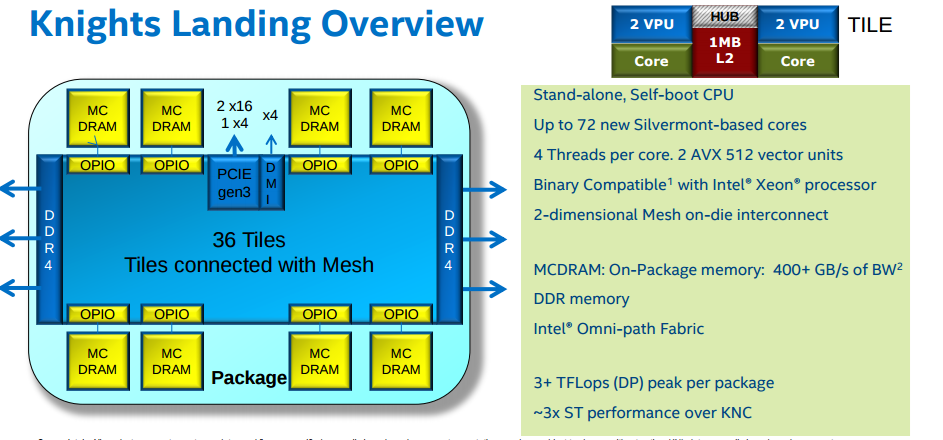
KNL is the evolution of Knights Corner (KNC), available on GALILEO until January 2018. The main differences between KNC and KNL are:
- KNLs are standalone, self-bootable processors, unlike KNCs, which are treated as accelerators;
- a better power in performance;
- a faster internal network.
Applications compiled for KNL are binary compatible with regular computing nodes.
KNL supports Intel AVX-512 instruction set extensions. The same three login nodes serve the Marconi-Broadwell (Marconi-A1) and the Marconi-KNL (Marconi-A2) partitions and queueing systems.
Storage devices are in common between the two partitions.

This supercomputer is available to European researchers as a Tier-0 system of the PRACE (www.prace-project.eu) infrastructure, as well as to Italian public and industrial researchers.
Part of this system (MARCONI_Fusion) is reserved for the activity of EUROfusion (https://www.euro-fusion.org/). Details on the MARCONI_Fusion environment are reported in a dedicated document.
Access
All the login nodes have an identical environment and can be reached with SSH (Secure Shell) protocol using the "collective" hostname:
> login.marconi.cineca.it
which establishes a connection to one of the available login nodes.
For information about data transfer from other computers please follow the instructions and caveats on the dedicated section Data storage, or the document Data Management.
Accounting
For accounting information please consult our dedicated section.
The account_no (or project) is important for batch executions. You need to indicate an account_no to be accounted for in the scheduler, using the flag "-A"
#SBATCH -A <account_no>
Please remember that different projects are usually active on different hosts. With the "saldo -b" command you can list all the account_no associated to your username. On systems like MARCONI, where different partitions are available on the same environment, you need to specify the host name in the "saldo" command:
> saldo -b (reports projects defined on Marconi-KNL - default)
> saldo -b --knl (reports projects defined on Marconi-KNL)
> saldo -b --skl (reports projects defined on Marconi-SKL)
Budget Linearization policy
On MARCONI a linearization policy for the usage of project budgets has been defined and implemented. For each account, a monthly quota is defined as:
monthTotal = (total_budget / total_no_of_months)
Starting from the first day of each month, the collaborators of any account are allowed to use the quota at full priority. As long as the budget is consumed, the jobs submitted from the account will gradually lose priority, until the monthly budget (monthTotal) is fully consumed. At that moment, their jobs will still be considered for execution, but with a lower priority than the jobs from accounts that still have some monthly quota left.
This policy is similar to those already applied by other important HPC centers in Europe and worldwide. The goal is to improve the response time, giving users the opportunity of using the cpu hours assigned to their project in relation of their actual size (total amount of core-hours).
Disks and Filesystems
The storage organization conforms to the CINECA infrastructure (see Section Data Storage and Filesystems).
In addition to the home directory $HOME, for each user is defined a scratch area $CINECA_SCRATCH, a large disk for the storage of run time data and files.
A $WORK area is defined for each active project on the system, reserved for all the collaborators of the project. This is a safe storage area to keep run time data for the whole life of the project.
| Total Dimension (TB) | Quota (GB) | Notes | |
|---|---|---|---|
| $HOME | 200 | 50 |
|
| $CINECA_SCRATCH | 2.500 | no quota |
|
| $WORK | 7.100 | 1.000 |
|
$DRES environment variable points to the shared repository where Data RESources are maintained. This is a data archive area available only on-request, shared with all CINECA HPC systems and among different projects. $DRES is not mounted on the compute nodes. This means that you cannot access it within a batch job: all data needed during the batch execution has to be moved to $WORK or $CINECA_SCRATCH before the run starts.
Since all the filesystems are based on IBM Spectrum Scale™ file system (formerly GPFS), the usual unix command "quota" is not working. Use the local command cindata to query for disk usage and quota ("cindata -h" for help):
> cindata
Modules environment
As usual, the software modules are collected in different profiles and organized by functional category (compilers, libraries, tools, applications,..).
On Marconi a new feature has been added to the module environment: the profiles are of two types, “domain” type (chem, phys, lifesc,..) for the production activity and “programming” type (base and advanced) for compilation, debugging and profiling activities and that they can be loaded together.
Applications available on MARCONI are compiled for A1-A3 and A2 partitions. At present we did not recompile all software to have a KNL optimized version, but the binaries built for the Broadwell nodes can run on the KNL as well.
"Base" profile is the default. It is automatically loaded after login and it contains basic modules for the programming activities (intel e gnu compilers, math libraries, profiling and debugging tools,..).
If you want to use a module placed under others profiles, for example an application module, you will have to load the corresponding profile in advance:
>module load profile/<profile name>
>module load autoload <module name>
For listing all profiles you have loaded use the following command:
>module list
In order to detect all profiles, categories and modules available on Marconi the command “modmap” is available:
>modmap
Production environment
Since MARCONI is a general purpose system and it is used by several users at the same time, long production jobs must be submitted using a queuing system. This guarantees that the access to the resources is as fair as possible.
Roughly speaking, there are two different modes to use an HPC system: Interactive and Batch. For a general discussion see the section Production Environment and Tools.
Interactive
A serial program can be executed in the standard UNIX way:
> ./program
This is allowed only for very short runs, since the interactive environment has a 10 minutes time limit: for longer runs please use the "batch" mode.
A parallel program can be executed interactively only within an "Interactive" SLURM batch job, using the "srun" command: the job is queued and scheduled as any other job, but when executed, the standard input, output, and error streams are connected to the terminal session from which srun was launched.
For example, to start an interactive session with the MPI program myprogram, using one node, two processors, launch the command:
> srun -N1 -n2 --ntasks-per-node=2 -A <account_name> --pty /bin/bash
SLURM will then schedule your job to start, and your shell will be unresponsive until free resources are allocated for you.
When the shell come back with the prompt, you can execute your program by typing:
> srun ./myprogram
or
> mpirun ./myprogram
The srun command will take by default PMI2 as MPI type.
Please note that
1) The recommended way to launch parallel tasks in slurm jobs is with srun. By using srun vs mpirun you will get full support for process tracking, accounting, task affinity, suspend/resume and other features.
2) Controlling the processes and threads affinity is crucial to ensure the optimal performances on Marconi-A2 and Marconi-A3. Do not rely on slurm autoaffinity and use the proper SLURM --cpu-bind option
SLURM automatically exports the environment variables you defined in the source shell, so that if you need to run your program myprogram in a controlled environment (i.e. specific library paths or options), you can prepare the environment in the origin shell being sure to find it in the interactive shell.
Batch
The info reported here refer to the general MARCONI partition. The production environment of MARCONI_Fusion is discussed in a separate document.
As usual on systems using SLURM, you can submit a script script.x using the command:
> sbatch script.x
You can get a list of defined partitions with the command:
> sinfo
You can simplify the output reported by the sinfo command specifying the output format via the "-o" option. A minimal output is reported, for instance, with:
> sinfo -o "%10D %20F %P"
which shows, for each partition, the total number of nodes and the number of nodes by state in the format "Allocated/Idle/Other/Total".
IMPORTANT:
- Please note that the recommended way to launch parallel tasks in slurm jobs is with srun. By using srun vs mpirun you will get full support for process tracking, accounting, task affinity, suspend/resume and other features.
- Controlling the processes and threads affinity is crucial to ensure the optimal performances on Marconi-A2 and Marconi-A3. Do not rely on slurm autoaffinity and use the proper SLURM --cpu-bind option.
For more information and examples of job scripts, see section Batch Scheduler SLURM.
Submitting serial Batch jobs
The bdw_all_serial partition is available with a maximum walltime of 4 hours, 6 tasks and 18000 MB per job. It runs on two dedicated nodes, and it is designed for pre/post-processing serial analysis, and for moving your data (via rsync, scp etc.) in case more than 10 minutes are required to complete the data transfer. In order to use this partition you have to specify the SLURM flag "-p":
#SBATCH -p bdw_all_serial
Submitting Batch jobs for A2 partition
On MARCONI it is possible to submit jobs requiring different resources by specifying the corresponding partition. If you do not specify the partition, your jobs will run on the bdw_all_serial partition.
If you do not specify the walltime (by means of the #SBATCH --time directive), a default value of 30 minutes will be assumed.
[username@r000u07l02 ~]$
With respect to the previous configuration. submission process results simplified. You do not more need to load the specific "env-knl" module to submit jobs on partitions based on Knights Landing processors. Instead, you simply need to specify the correct partition using the "SBATCH -p directive. Choosing a knl_***_*** partition, you will be sure to work on KNL nodes.
Each KNL node exposes itself to SLURM as having 68 cores (corresponding to the physical cores of the KNL processor). Jobs should request the entire node (hence, #SBATCH -n 68), and the KNL SLURM server is configured so that to assign the KNL nodes in an exclusive way (even if less ncpus are asked). Hyper-threading is enabled, hence you can run up to 272 processes/threads on each assigned node.
The configuration of the Marconi-A2 partition allowed to require different HBM modes (on-package high-bandwidth memory based on the multi-channel dynamic random access memory (MCDRAM) technology) and clustering modes of cache operations:
#SBATCH --constraint=flat/cache
Please refer to the official Intel documentation for a description of the different modes.
For the queues serving the Marconi FUSION partition, please refer to the dedicated document.
The maximum memory which can be requested is 86000MB for cache/flat nodes:
#SBATCH --mem=86000
(the default measurement unity, if not specified, is MB)
For flat nodes the jobs can require the KNL high bandwidth memory (HBM) using Slurm's Generic RESource (GRES) options:
#SBATCH --constraint=flat
#SBATCH --mem=86000
For example, to request a single KNL node in a production queue the following SLURM job script can be used:
#!/bin/bash
#SBATCH -N 1
#SBATCH -A <account_name>
#SBATCH --mem=86000
#SBATCH -p knl_usr_prod
#SBATCH --time 00:05:00
#SBATCH --job-name=KNL_batch_job
#SBATCH --mail-type=ALL
#SBATCH --mail-user=<user_email>
srun ./myexecutable
where <account_name> is the project account user has to specify to pay for the job, and <user_email> is a valid email address user can specify to receive real-time information about his job status changes.
Submitting Batch jobs for A3 partition
Most of the A3-SkyLake nodes are reserved for EuroFusion users only.
sinfo -d lists the following partitions.
skl_fua_prod and skl_fua_dbg, as it is obvious, are reserved to EuroFusion users.
skl_usr_skl and skl_usr_dbg are, instead, opened to academic production.
Each SKL node exposes itself to SLURM as having 48 cores (corresponding to the 48 physical cores of the SKL processor). Jobs should request the entire node (hence, ncpus=48).
As for KNL partition, SLURM assigns a SkyLake node in exclusive way, i.e. user will pay for full node even if only requests 1 task per node.
The maximum memory which can be requested is 182000MB and this value guarantees that no memory swapping will occur.
For example, to request a single SKL node in a production queue the following SLURM job script can be used:
#!/bin/bash
#SBATCH -N 1
#SBATCH -A <account_name>
#SBATCH --mem=180000
#SBATCH -p skl_usr_prod
#SBATCH --time 00:05:00
#SBATCH --job-name=SKL_batch_job
#SBATCH --mail-type=ALL
#SBATCH --mail-user=<user_email>
srun ./myexecutable
The QOS qos_lowprio is automatically associated to users with exhausted but still active project; the QOS allows those users to keep using the SKL resources but at zero priority.
Summary
In the following table you can find all the main features and limits imposed on the queues of the shared A1 and A2 partitions. For Marconi-FUSION dedicated queues please refer to the dedicated document.
MARCONI Partition | SLURM partition | QOS | # cores per job | max walltime | max running jobs per user/ max n. of cpus/nodes per user | max memory per node (MB) | priority | HBM/clustering mode | notes |
front-end | bdw_all_serial (default partition) | noQOS | max = 6 (max mem= 18000 MB) | 04:00:00 | 6 cpus | 18000 | 40 | ||
| A1 | qos_rcm | min = 1 max = 48 | 03:00:00 | 1/48 | 182000 | - | runs on 24 nodes shared with the debug queue on SKL | ||
| A2 | knl_usr_dbg | no QOS | min = 1 node max = 2 nodes | 00:30:00 | 5/5 | 86000 (cache) | 40 | runs on 144 dedicated nodes | |
| A2 | knl_usr_prod | no QOS | min = 1 node max = 195 nodes | 24:00:00 | 1000 nodes | 86000 (cache) | 40 | ||
| knl_qos_bprod | min = 196 nodes max = 1024 nodes | 24:00:00 | 1/1000 | 86000 (cache) | 85 | #SBATCH -p knl_usr_prod #SBATCH --qos=knl_qos_bprod | |||
| qos_special | >1024 nodes | >24:00:00 (max = 195 nodes for user) | 86000 (cache) | 40 | #SBATCH --qos=qos_special request to superc@cineca.it | ||||
| A3 | skl_usr_dbg | no QOS | min = 1 node max = 4 nodes | 00:30:00 | 4/4 | 182000 | 40 | runs on 24 dedicated nodes | |
| A3 | skl_usr_prod | no QOS | min = 1 node max = 64 nodes | 24:00:00 | 64 nodes | 182000 | 40 | ||
| skl_qos_bprod | min=65 max = 256 | 24:00:00 | 1/256 1 jobs per account | 182000 | 85 | #SBATCH -p skl_usr_prod #SBATCH --qos=skl_qos_bprod | |||
| qos_special | >256 nodes | >24:00:00 (max = 64 nodes for user) | 182000 | 40 | #SBATCH --qos=qos_special request to superc@cineca.it | ||||
| qos_lowprio | max = 64 nodes | 24:00:00 | 64 nodes | 182000 | 0 | #SBATCH --qos=qos_lowprio |
Graphic session
If a graphic session is desired we recommend to use the tool RCM (Remote Connection Manager)_OLD. For additional information visit Remote Visualization section on our User Guide.
Programming environment
The programming environment of the MARCONI cluster consists of a choice of compilers for the main scientific languages (Fortran, C and C++), debuggers to help users in finding bugs and errors in the codes, profilers to help in code optimisation.
In general you must "load" the correct environment also for using programming tools like compilers, since "native" compilers are not available.
If you use a given set of compilers and libraries to create your executable, very probably you have to define the same "environment" when you want to run it. This is because, since by default linking is dynamic on Linux systems, at runtime the application will need the compiler shared libraries as well as other proprietary libraries. This means that you have to specify "module load" for compilers and libraries, both at compile time and at run time. To minimize the number of needed modules at runtime, use static linking to compile the applications.
Compilers
You can check the complete list of available compilers on MARCONI with the command:
> module available
and checking the "compilers" section. In general, the available compilers are:
INTEL (ifort, icc, icpc) :
> module load intel
GNU (gcc, g77, g95):
> module load gnu
PGI - Portland Group (pgf77,pgf90,pgf95,pghpf, pgcc, pgCC):
> module load profile/advanced
> module load pgi
After loading the appropriate module, use the "man" command to get the complete list of the flags supported by the compiler, for example:
> module load intel
> man ifort
There are some flags that are common for all these compilers. Others are more specifics. The most common are reported later for each compiler.
- If you want to use a specific library or a particular include file, you have to give their paths, using the following options
-I/path_include_files specify the path of the include files
-L/path_lib_files -l<xxx> specify a library lib<xxx>.a in /path_lib_files
2. If yout want to debug your code you have to turn off optimization and turn on run time checkings: these flags are described in the following section.
If you want to compile your code for normal production you have to turn on optimization by choosing a higher optimization level:
-O2 or -O3 Higher optimization levels
Other flags are available for specific compilers and are reported later.
INTEL Compilers
Initialize the environment with the module command:
> module load intel
The names of the Intel compilers are:
- ifort: Fortran77 and Fortran90 compiler
- icc: C compiler
- icpc: C++ compiler
The documentation can be obtained with the man command after loading the relevant module:
> man ifort > man icc
Some miscellanous flags are described in the following:
-extend_source Extend over the 77 column F77's limit -free / -fixed Free/Fixed form for Fortran -ip Enables interprocedural optimization for single-file compilation -ipo Enables interprocedural optimization between files - whole program optimisation
Compiling for KNL and SKL
Since KNL and SKL nodes are binary compatible with legacy x86 instruction set, any code compiled for BDW nodes will run on these nodes. However, specific compiler option is needed to generate AVX-512 instructions to derive better performance from these nodes.
Version 15.0 and newer of the Intel compilers can generate these instructions if you specify for KNL nodes the "-xMIC-AVX512" flag (which generates specific AVX512 instructions, hence the binary will not work on the Broadwell partition) or the -axMIC-AVX512 flag (which generates optimized executables for both AVX2 and AVX512 ISA):
module load intel
icc -axMIC-AVX512 -O3 -o executable source.c
icpc -axMIC-AVX512 -O3 -o executable source.cc
ifort -axMIC-AVX512 -O3 -o executable source.f
Differently for SKL nodes you have to specify the -"xCORE-AVX512" flag in order to generate AVX-512 instructions. When using this option, Intel compilers default to using AVX512 “low”, i.e., a 256-bit version of AVX512 through AVX512-VL (see also compiler documentation for -qopt-zmm-usage=low).
This means that by default, the compiler generates instructions which operate only on 256 bits of the 512 bit registers, but benefit from things like masking and doubled register set size. The most obvious benefit from this approach is that the frequency drop equals only that of AVX2 code.
If you have a code which vectorizes well, you can try experimenting with "-xCORE-AVX512 -qopt-zmm-usage=high" to make the compiler generate AVX-512 code with full 512 bit vectors in use. Doing this will cause the clock frequency of the CPU to drop rather significantly which in turn often causes the code either to run slower or at equal speed compared to for instance AVX2.”
There are certain considerations to be taken into account before running legacy codes on KNL and SKL nodes. Primarily, the effective use of vector instructions is critical to achieve good performance on the cores. For guideline on how to get vectorization information and improve code vectorization, refer to
How to Improve Code Vectorization
PORTLAND Group (PGI)
Initialize the environment with the module command:
> module load profile/advanced > module load pgi
The name of the PGI compilers are:
- pgf77: Fortran77 compiler
- pgf90: Fortran90 compiler
- pgf95: Fortran95 compiler
- pghpf: High Performance Fortran compiler
- pgcc: C compiler
- pgCC: C++ compiler
The documentation can be obtained with the man command after loading the relevant module:
> man pgf95 > man pgcc
Some miscellanous flags are described in the following:
-Mextend To extend over the 77 column F77's limit -Mfree / -Mfixed Free/Fixed form for Fortran -fast Chooses generally optimal flags for the target platform -fastsse Chooses generally optimal flags for a processor that supports SSE instructions
> man gcc
Some miscellanous flags are described in the following:
-ffixed-line-length-132 To extend over the 77 column F77's limit -ffree-form / -ffixed-form Free/Fixed form for Fortran
GNU compilers
The gnu compilers are always available but they are not the best optimizing compilers. You do not need to load the module for using them.
The name of the GNU compilers are:
- g77: Fortran77 compiler
- gfortran: Fortran95 compiler
- gcc: C compiler
- g++: C++ compiler
The documentation can be obtained with the man command:
> man gfortan
Debuggers and Profilers
If at runtime your code dies, then there is a problem. In order to solve it, you can decide to analyze the core file (core not available with PGI compilers) or to run your code using the debugger.
Compiler flags
Whatever your decision, in any case you need to enable compiler runtime checks, by putting specific flags during the compilation phase. In the following we describe those flags for the different Fortran compilers: if you are using the C or C++ compiler, please check before because the flags may differ.
The following flags are generally available for all compilers and are mandatory for an easier debugging session:
-O0 Lower level of optimization -g Produce debugging information
Other flags are compiler specific and are described in the following:
INTEL Fortran compiler
The following flags are useful (in addition to "-O0 -g") for debugging your code:
-traceback generate extra information to provide source file traceback at run time -fp-stack-check generate extra code to ensure that the floating-point stack is in the expected state -check bounds enables checking for array subscript expressions -fpe0 allows some control over floating-point exception handling at run-time
PORTLAND Group (PGI) Compilers
The following flags are useful (in addition to "-O0 -g") for debugging your code:
-C Add array bounds checking
-Ktrap=ovf,divz,inv Controls the behavior of the processor when exceptions occur:
FP overflow, divide by zero, invalid operands
GNU Fortran compilers
The following flags are useful (in addition to "-O0 -g")for debugging your code:
-Wall Enables warnings pertaining to usage that should be avoided -fbounds-check Checks for array subscripts.
Debuggers available
Totalview
NOTE: if debugging with Intel compiler suite, you may want to add in your jobscript the following line, before the execution of TotalView:
source $INTELMPI_HOME/bin64/mpivars.sh debug
to provide a better population of the message queue.
Scalasca
....
In the following we report information about other ways to debug your codes:
PGI: pgdbg (serial/parallel debugger)
pgdbg is the Portland Group Inc. symbolic source-level debugger for F77, F90, C, C++ and assembly language programs. It is capable of debugging applications that exhibit various levels of parallelism, including:
- Single-thread, serial applications
- Multi-threaded applications
- Distributed MPI applications
- Any combination of the above
There are two forms of the command used to invoke pgdbg. The first is used when debugging non-MPI applications, the second form, using mpirun, is used when debugging MPI applications:
> pgdbg [options] ./myexec [args] > mpirun [options] -dbg=pgdbg ./myexec [args]
More details in the on line documentation, using the "man pgdbg" command after loading the module.
To use this debugger, you should compile your code with one of the pgi compilers and the debugging command-line options described above, then you run your executable inside the "pgdbg" environment:
> module load pgi > pgf90 -O0 -g -C -Ktrap=ovf,divz,inv -o myexec myprog.f90 > pgdbg ./myexec
By default, pgdbg presents a graphical user interface (GUI). A command-line interface is also provided though the "-text" option.
GNU: gdb (serial debugger)
GDB is the GNU Project debugger and allows you to see what is going on 'inside' your program while it executes -- or what the program was doing at the moment it crashed.
GDB can do four main kinds of things (plus other things in support of these) to help you catch bugs in the act:
- Start your program, specifying anything that might affect its behavior.
- Make your program stop on specified conditions.
- Examine what has happened, when your program has stopped.
- Change things in your program, so you can experiment with correcting the effects of one bug and go on to learn about another.
More details in the on line documentation, using the "man gdb" command.
To use this debugger, you should compile your code with one of the gnu compilers and the debugging command-line options described above, then you run your executable inside the "gdb" environment:
> gfortran -O0 -g -Wall -fbounds-check -o myexec myprog.f90 > gdb ./myexec
VALGRIND
Valgrind is a framework for building dynamic analysis tools. There are Valgrind tools that can automatically detect many memory management and threading bugs, and profile your programs in detail. The Valgrind distribution currently includes six production-quality tools: a memory error detector, two thread error detectors, a cache and branch-prediction profiler, a call-graph generating cache profiler, and a heap profiler.
Valgrind is Open Source / Free Software, and is freely available under the GNU General Public License, version 2.
To analyze a serial application:
- Load Valgrind module --> module load valgrind
- Load module for the compiler and compile your code with the compiler you prefer (Use -O0 -g flags)
- Run the executable under Valgrind.
If you normally run your program like this:
myprog arg1 arg2
Use this command line:
valgrind (valgrind-options) myprog arg1 arg2
Memcheck is the default tool. You can add the --leak-check option that turns on the detailed memory leak detector. Your program will run much slower than normal, and use a lot more memory. Memcheck will issue messages about memory errors and leaks that it detects.
- Load Valgrind module --> module load valgrind
- Load modules for compiler and openmpi libraries (at present only available for intel and gnu)
- Compile your code with the "-O0 -g" flags both at compiling and linking time
- Run the executable under Valgrind (both in interactive than in bacth mode)
mpirun -np 4 valgrind (valgrind-options) myprog arg1 arg2
Core file analisys
In order to understand what problem was affecting you code, you can also try a "Core file" analisys. Since core files are usually quite large, be sure to work in the /scratch area.
There are several steps to follow:
- Increase the limit for possible core dumping
> ulimit -c unlimited (bash) > limit coredumpsize unlimited (csh/tcsh)
- If you are using Intel compilers, set to TRUE the decfort_dump_flag environment variable
> export decfort_dump_flag=TRUE (bash) > setenv decfort_dump_flag TRUE (csh/tcsh)
- Compile your code with the debug flags described above.
- Run your code and create the core file.
- Analyze the core file using different tools depending on the original compiler.
INTEL compilers
> module load intel > ifort -O0 -g -traceback -fp-stack-check -check bounds -fpe0 -o myexec prog.f90 > ulimit -c unlimited > export decfort_dump_flag=TRUE > ./myexec > ls -lrt -rwxr-xr-x 1 aer0 cineca-staff 9652 Apr 6 14:34 myexec -rw------- 1 aer0 cineca-staff 319488 Apr 6 14:35 core.25629 > idbc ./myexec core.25629
PGI compilers
> module load pgi > pgf90 -O0 -g -C -Ktrap=ovf,divz,inv -o myexec myprog.f90 > ulimit -c unlimited > ./myexec > ls -lrt -rwxr-xr-x 1 aer0 cineca-staff 9652 Apr 6 14:34 myexec -rw------- 1 aer0 cineca-staff 319488 Apr 6 14:35 core.25666 > pgdbg -text -core core.25666 ./myexec
GNU Compilers
> gfortran -O0 -g -Wall -fbounds-check -o myexec prog.f90 > ulimit -c unlimited > ./myexec > ls -lrt -rwxr-xr-x 1 aer0 cineca-staff 9652 Apr 6 14:34 myexec -rw------- 1 aer0 cineca-staff 319488 Apr 6 14:35 core.25555 > gdb ./myexec core.2555
Profilers (gprof)
In software engineering, profiling is the investigation of a program's behavior using information gathered as the program executes. The usual purpose of this analisys is to determine which sections of a program to optimize - to increase its overall speed, decrease its memory requirement or sometimes both.
A (code) profiler is a performance analisys tool that, most commonly, measures only the frequency and duration of function calls, but there are other specific types of profilers (e.g. memory profilers) in addition to more comprehensive profilers, capable of gathering extensive performance data.
gprof
The GNU profiler gprof is a useful tool for measuring the performance of a program. It records the number of calls to each function and the amount of time spent there, on a per-function basis. Functions which consume a large fraction of the run-time can be identified easily from the output of gprof. Efforts to speed up a program should concentrate first on those functions which dominate the total run-time.
gprof uses data collected by the -pg compiler flag to construct a text display of the functions within your application (call tree and CPU time spent in every subroutine). It also provides quick access to the profiled data, which let you identify the functions that are the most CPU-intensive. The text display also lets you manipulate the display in order to focus on the application's critical areas.
Usage:
> gfortran -pg -O3 -o myexec myprog.f90 > ./myexec > ls -ltr ....... -rw-r--r-- 1 aer0 cineca-staff 506 Apr 6 15:33 gmon.out > gprof myexec gmon.out
It is also possible to profile at code line-level (see "man gprof" for other options). In this case you must use also the “-g” flag at compilation time:
> gfortran -pg -g -O3 -o myexec myprog.f90 > ./myexec > ls -ltr ....... -rw-r--r-- 1 aer0 cineca-staff 506 Apr 6 15:33 gmon.out > gprof -annotated-source myexec gmon.out
It is possilbe to profile MPI programs. In this case the environment variable GMON_OUT_PREFIX must be defined in order to allow to each task to write a different statistical file. Setting
export GMON_OUT_PREFIX=<name>
once the run is finished each task will create a file with its process ID (PID) extension
<name>.$PID
If the environmental variable is not set every task will write the same gmon.out file.
Intel VTUNE Amplifier
Intel performance analisys toolkit that can be used to identify bottlenecks in an application. The tool can be used to perform different types of analisys.
To start the analisys use the command line interface:
module load autoload vtune
amplxe-cl -collect hotspots -r <application_path> <vtune_options> <application>
To explore the results of the analisys performed use the Intel VTune Amplifier GUI:
amplxe-gui <application_path>
Please note that to get a correct result in terms of CPU time it is suggested to use of the advanced-hotspots analisys on both Marconi KNL and SKL partitions. For example:
amplxe-cl -collect advanced-hotspots --target-duration-type veryshort <executable>
Scientific libraries
MKL
The Intel Math Kernel Library (Intel MKL) enables improving performance of scientific, engineering, and financial software that solves large computational problems. Intel MKL provides a set of linear algebra routines, fast Fourier transforms, as well as vectorized math and random number generation functions, all optimized for the latest Intel processors, including processors with multiple cores.
Intel MKL is thread-safe and extensively threaded using the OpenMP technology
documentation can be found in:
${MKLROOT}/../Documentation/en_US/mkl
To use the MKL in your code you to load the module, then to define includes and libraries at compile and linking time:
> module load mkl > icc -I$MKL_INC -L$MKL_LIB -lmkl_intel_lp64 -lmkl_core -lmkl_sequential
For more inormation please refer to the documentation.
Parallel programming
The parallel programming on MARCONI is based on IntelMPI and OpenMPI versions of MPI. The libraries and special wrappers to compile and link the personal programs are contained in several modules, one for each supported suite of compilers.
The main four parallel-MPI commands for compilation are:
- mpiifort (IntelMPI)/ mpif90 (OpenMPI) (Fortran90)
- mpiifort (IntelMPI) / mpif77 (OpenMPI) (Fortran77)
- mpiicc (IntelMPI) / mpicc (OpenMPI) (C)
- mpiicpc (IntelMPI) / mpicxx (C++)
These command names refers to wrappers around the actual compilers, they behave differently depending on the module you have loaded.
> module avail intelmpi intelmpi/2017--binary(default) intelmpi/5.1--binary > module load intel/pe-xe-2017--binary intelmpi/2017--binary > man mpiifort > mpiifort -o myexec myprof.f90 (uses the ifort compiler)
Please note that the Intel mpif90 wrapper is meant for using the GNU gfortran compiler instead of Intel ifort. If you need to use the IntelMPI libraries with the GNU gfortran compiler you need to redefine the I_MPI_F90 variable:
> module load autoload intelmpi/2017–binary
> export I_MPI_F90=gfortran
> mpif90 -o myexec myprof.f90 (uses the gfortran compiler)
With the above setting the system gfortran (4.8) compiler will be used. You can use a more recent version loading the gnu/6.1.0 module together with the intelmpi/2017–binary.
> module avail openmpi openmpi/1-10.3--gnu--6.1.0
> module load gnu openmpi/1-10.3--gnu--6.1.0 > man mpif90 > mpif90 -o myexec myprof.f90 (uses the gfortran compiler)
The parallel applications have to be executed with the command:
> mpirun ./myexec
There are limitations on running parallel programs in the login shell. You should use the "Interactive SLURM" mode, as described in the "Interactive" section, previously in this page.
-Wall Enables warnings pertaining to usage that should be avoided -fbounds-check Checks for array subscripts.